{
"metadata": {
"name": "",
"signature": "sha256:41cfc86b9e6591b6c5c0923993a6474486bd348aee8984369117217bfef4980a"
},
"nbformat": 3,
"nbformat_minor": 0,
"worksheets": [
{
"cells": [
{
"cell_type": "heading",
"level": 1,
"metadata": {},
"source": [
"Problem Set 1 - DNA"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"**Total points:** 30\n",
"\n",
"**Due:** Wednesday 25th March 19:00\n",
"\n",
"**Format:** IPython Notebook\n",
"\n",
"*The number of points in this problem sheet is *not* directly proportional to the difficulty. In fact, Part 3 is more difficult than Part 1 and 2, but is worth fewer points in terms of the amount of work/code, and are there for people who enjoy challenges :) So you can choose to not complete Part 3, and you should still be able to get more than 60% of points from the previous questions if you answer them correctly.*"
]
},
{
"cell_type": "heading",
"level": 2,
"metadata": {},
"source": [
"Background"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
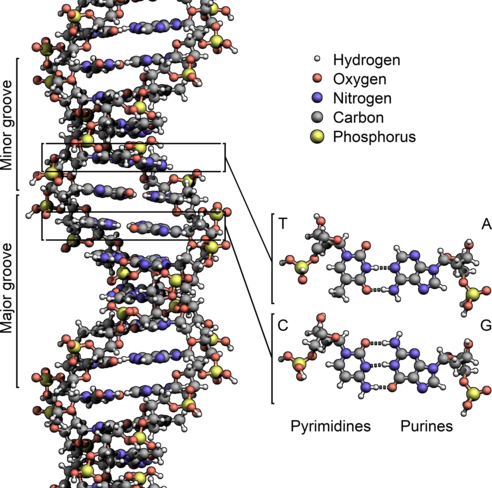
"This problem set is about DNA data and methods typically applied to this kind of data. As a reminder, [DNA](http://en.wikipedia.org/wiki/DNA) is a molecule that encodes genetic instructions for living organisms. DNA is typically found as a double-stranded helix, in which each strand corresponds to a sequence of *nucleotides*. Each nucleotide consists of a nucleobase (guanine, adenine, thymine, or cytosine) attached to sugars, which are in turn separated from each other by phosphate groups:\n",
"\n",
"
\n",
"\n",
"\n",
"
\n",
"
\n",
"\n",
"(image from Wikipedia)\n",
"\n",
"The nucleobases are commonly referred to with the letters G (guanine), A (adenine), T (thymine), and C (cytosine). The nucleobases form pairs between the two strands: G pairs with C, and T pairs with A.\n",
"\n",
"DNA is therefore most commonly represented as a sequence of nucleobases, such as ``GATTACACCTCATTATAAA``.\n",
"\n",
"For more information about DNA, see the wikipedia page in [German](http://de.wikipedia.org/wiki/Desoxyribonukleins\u00e4ure) or [English](http://en.wikipedia.org/wiki/DNA).\n",
"\n",
"In this problem set, we will be working with fake genetic data, since real-life data often has added complications, but the functions and techniques uses here are the same or very similar to techniques used on real data."
]
},
{
"cell_type": "heading",
"level": 2,
"metadata": {},
"source": [
"Part 1 (10 points)"
]
},
{
"cell_type": "heading",
"level": 3,
"metadata": {},
"source": [
"Question 1 (6 points)"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"The **reverse complement** of DNA is found by reversing the DNA sequence, then replacing each base by its complement (A is replaced by T, T is replaced by A, G is replaced by C, and C is replaced by G). For example, the reverse complement of ``ATGCGGC`` is ``GCCGCAT``\n",
"\n",
"Write a Python function ``reverse_complement`` that takes a DNA sequence as a string, and returns the reverse complement. Test your function by ensuring that ``reverse_complement('ATGCGGC')`` is ``'GCCGCAT'``, then find the reverse complement of the following sequence:\n",
"\n",
" ATGCGCGGATCGTACCTAATCGATGGCATTAGCCGAGCCCGATTACGC\n",
" \n",
"Print the result out using ``print``.\n",
"\n",
"Note that this function is **NOT** needed for the remaining questions below."
]
},
{
"cell_type": "heading",
"level": 3,
"metadata": {},
"source": [
"Question 2 (4 points)"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"[Ribonucleic acid (RNA)](http://en.wikipedia.org/wiki/RNA) is a family of large biological molecules that is *transcribed* from DNA by the [RNA Polymerase](http://en.wikipedia.org/wiki/RNA_polymerase) enzyme. It consists of a single strand of nucleotides that are identical to the ones found in DNA, with the exception of uracil (U), which replaces thymine (T).\n",
"\n",
"[Messenger RNA](http://en.wikipedia.org/wiki/Messenger_RNA) molecules (or mRNA) are a subset of RNA molecules that are used to pass information from DNA to [ribosomes](http://en.wikipedia.org/wiki/Ribosome), which then translates the mRNA to protein sequences.\n",
"\n",
"Write a Python function ``dna_to_mrna`` that takes a DNA sequence and returns the corresponding mRNA sequence. For example, the DNA sequence ``ATCGCGAT`` should produce the mRNA sequence ``AUCGCGAU`` (note that you do **not** need to find the reverse complement of the DNA here)"
]
},
{
"cell_type": "heading",
"level": 2,
"metadata": {},
"source": [
"Part 2 (15 points)"
]
},
{
"cell_type": "heading",
"level": 3,
"metadata": {},
"source": [
"Question 3 (8 points)"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"When the mRNA is translated to a protein sequence, each set of three nucleotides, called a **codon**, is translated into a single amino acid. For example, the codon ``UUC`` translates to the amino acid *Phenylalanine*. Each amino acid can be represented by a single letter - for example Phenylalanine is represented by the letter ``F``. A protein, which is formed from a sequence of amino acids, can therefore be written as a sequence of letters in the same way as DNA or mRNA, but using more of the letters of the alphabet since there are more than four amino acids.\n",
"\n",
"The [data/problem_1_codons.txt](data/problem_1_codons.txt) file contains two columns. The first column gives a list of codons, and the second column gives the corresponding amino acid (represented by a single letter). Certain codons do not correspond to an amino acid, but instead indicate that the amino acid sequence is finished. These are indicated by ``Stop``.\n",
"\n",
"Write a function ``mrna_to_protein`` that takes an mRNA sequence (as a string) and returns the sequence of amino acids (as a string), stopping the first time a ``Stop`` codon is encountered. Make sure that the file is only read once when running the script (and not every time you want to translate a codon). You will likely need to use a Python dictionary to help.\n",
"\n",
"Finally, write a function ``dna_to_protein`` that takes a DNA sequence (as a string) and returns the sequence of amino acids (as a string), making use of the functions that you wrote previously.\n",
"\n",
"Print out the amino acid sequence for the following DNA sequence:\n",
"\n",
" AATCTCTACGGAAGTAGGTCAGTACTGATCGATCAGTCGATCGGGCGGCGATTTCGATCTGATTGTACGGCGGGCTAG\n",
" "
]
},
{
"cell_type": "heading",
"level": 3,
"metadata": {},
"source": [
"Question 4 (7 points)"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"In the previous questions, we have been specifying the DNA sequence by hand, but DNA sequences are usually long and are stored in files. A common file format is the [FASTA format](http://en.wikipedia.org/wiki/FASTA_format) which looks similar to this:\n",
"\n",
" >label1\n",
" ACTGTATCGATGCTAGCTACGTAGCTAGCTAGCTAGCTGACGTA\n",
" ACGATGTGCGAGGGTCATGGGACGCGAGCGAGTCTAGCACGATC\n",
" >label2\n",
" ACTGGGCTTGACTACGGCGGTATCTGACGGGCGAGCTGTACGAG\n",
" ACGGACTAGGGCGCGGCGGGGCGGATTTTCGAGTCGAGCGTTAT\n",
" \n",
"The first line starts with a ``>`` which is immediately followed by a label (which might be the name of the gene for example). The sequence then starts on the second line, and may continue on several lines. It is common to limit the length of each line to 80, but this may vary from file to file. The sequence stops once either the file ends, or a line starts with ``>``, which indicates that a new sequence is being given. There may be any number of sequences in a file.\n",
"\n",
"Write a function ``read_fasta``, that takes the name of a file (as a string) and returns a Python dictionary containing all the sequences from the file, with the keys in the dictionary corresponding to the label. If a sequence is given over several lines, you should remove any line returns and spaces. You should then be able to access the DNA for ``label1`` with ``d['label1']`` for example (if ``d`` is the name of the dictionary).\n",
"\n",
"Use this function and the functions you have written above to read in the [data/problem_1_question_4.fasta](data/problem_1_question_4.fasta) file and print out, for each sequence, the label, followed by the **amino acid** sequence (not the DNA sequence!)."
]
},
{
"cell_type": "heading",
"level": 2,
"metadata": {},
"source": [
"Part 3 (5 points)"
]
},
{
"cell_type": "heading",
"level": 3,
"metadata": {},
"source": [
"Question 5 (3 points)"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"Given several sequences with the same length, but which may include point mutations (i.e. individual nucleotides are changed), we want to try and find the most likely original sequence. For example, if we have the following sequences:\n",
"\n",
" sequence 1: A C T C T\n",
" sequence 2: A C T C G\n",
" sequence 3: G C C C T\n",
" sequence 2: A C T C T\n",
" sequence 4: A T G C T\n",
" \n",
"we can go through each position and find the most common nucleotide. To do this, we can first construct a matrix that looks like:\n",
" \n",
" A: 4 0 0 0 0\n",
" C: 0 4 1 5 0\n",
" G: 1 0 1 0 1\n",
" T: 0 1 3 0 4\n",
" \n",
"which indicates how many nucleotides of each type are found at each position, and from this we can see that the most common first base is A, the most common second base is C, and so on. The most common sequence is then ``ACTCT``. This is the [**consensus sequence**](http://en.wikipedia.org/wiki/Consensus_sequence).\n",
"\n",
"Write a function ``consensus_sequence`` that takes a dictionary of sequences (such as the one returned by ``read_fasta``), and computes then returns the consensus sequence. Read the [data/problem_1_question_5.fasta](data/problem_1_question_5.fasta) file using the function you wrote previsouly, and print out the corresponding consensus sequence. All the sequences in this file are the same length."
]
},
{
"cell_type": "heading",
"level": 3,
"metadata": {},
"source": [
"Question 6 (2 points)"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"In some cases, it is useful to be able to identify the longest common sub-sequence between two sequences. For example, in the sequences ``ACTGCT`` and ``TGCCCT``, the longest common sub-sequence is ``TGC`` (AC**TGC**T and **TGC**CCT). Note that these do not have to be at the same positon in each sequence.\n",
"\n",
"Write a function, ``longest_common_sequence``, that takes a dictionary of sequences (such as the one returned by ``read_fasta``) and returns the longest common sub-sequence found in all the sequences.\n",
"\n",
"Read the [data/problem_1_question_6.fasta](data/problem_1_question_6.fasta) file, and print out the longest common sequence between all the sequences."
]
}
],
"metadata": {}
}
]
}